Molpath Tumor Only Workflow¶
MolpathTumorOnlyWorkflow · 1 contributor · 1 version
No documentation was provided: contribute one
Quickstart¶
from janis_bioinformatics.tools.pmac.molpathTumorOnlyWorkflow import MolpathTumorOnly_1_0_0 wf = WorkflowBuilder("myworkflow") wf.step( "molpathtumoronlyworkflow_step", MolpathTumorOnly_1_0_0( sample_name=None, fastqs=None, seqrun=None, reference=None, region_bed=None, region_bed_extended=None, region_bed_annotated=None, genecoverage_bed=None, genome_file=None, panel_name=None, vcfcols=None, snps_dbsnp=None, snps_1000gp=None, known_indels=None, mills_indels=None, mutalyzer_server=None, pathos_db=None, maxRecordsInRam=None, gnomad=None, ) ) wf.output("fastq_qc", source=molpathtumoronlyworkflow_step.fastq_qc) wf.output("markdups_bam", source=molpathtumoronlyworkflow_step.markdups_bam) wf.output("doc_out", source=molpathtumoronlyworkflow_step.doc_out) wf.output("summary", source=molpathtumoronlyworkflow_step.summary) wf.output("gene_summary", source=molpathtumoronlyworkflow_step.gene_summary) wf.output("region_summary", source=molpathtumoronlyworkflow_step.region_summary) wf.output("gridss_vcf", source=molpathtumoronlyworkflow_step.gridss_vcf) wf.output("gridss_bam", source=molpathtumoronlyworkflow_step.gridss_bam) wf.output("haplotypecaller_vcf", source=molpathtumoronlyworkflow_step.haplotypecaller_vcf) wf.output("haplotypecaller_bam", source=molpathtumoronlyworkflow_step.haplotypecaller_bam) wf.output("haplotypecaller_norm", source=molpathtumoronlyworkflow_step.haplotypecaller_norm) wf.output("mutect2_vcf", source=molpathtumoronlyworkflow_step.mutect2_vcf) wf.output("mutect2_bam", source=molpathtumoronlyworkflow_step.mutect2_bam) wf.output("mutect2_norm", source=molpathtumoronlyworkflow_step.mutect2_norm) wf.output("addbamstats_vcf", source=molpathtumoronlyworkflow_step.addbamstats_vcf)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for MolpathTumorOnlyWorkflow:
# user inputs
janis inputs MolpathTumorOnlyWorkflow > inputs.yaml
inputs.yaml
fastqs:
- - fastqs_0.fastq.gz
- fastqs_1.fastq.gz
- - fastqs_0.fastq.gz
- fastqs_1.fastq.gz
genecoverage_bed: genecoverage_bed.bed
genome_file: genome_file.txt
gnomad: gnomad.vcf.gz
known_indels: known_indels.vcf.gz
maxRecordsInRam: 0
mills_indels: mills_indels.vcf.gz
mutalyzer_server: <value>
panel_name: <value>
pathos_db: <value>
reference: reference.fasta
region_bed: region_bed.bed
region_bed_annotated: region_bed_annotated.bed
region_bed_extended: region_bed_extended.bed
sample_name: <value>
seqrun: <value>
snps_1000gp: snps_1000gp.vcf.gz
snps_dbsnp: snps_dbsnp.vcf.gz
vcfcols: vcfcols.txt
- Run MolpathTumorOnlyWorkflow with:
janis run [...run options] \
--inputs inputs.yaml \
MolpathTumorOnlyWorkflow
Information¶
URL: No URL to the documentation was provided
| ID: | MolpathTumorOnlyWorkflow |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | v1.0.0 |
| Authors: | Jiaan Yu |
| Citations: | |
| Created: | 2020-06-12 |
| Updated: | 2020-08-10 |
Outputs¶
| name | type | documentation |
|---|---|---|
| fastq_qc | Array<Array<Zip>> | |
| markdups_bam | IndexedBam | |
| doc_out | TextFile | |
| summary | csv | |
| gene_summary | TextFile | |
| region_summary | TextFile | |
| gridss_vcf | VCF | |
| gridss_bam | BAM | |
| haplotypecaller_vcf | Gzipped<VCF> | |
| haplotypecaller_bam | IndexedBam | |
| haplotypecaller_norm | VCF | |
| mutect2_vcf | Gzipped<VCF> | |
| mutect2_bam | Optional<IndexedBam> | |
| mutect2_norm | VCF | |
| addbamstats_vcf | VCF |
Workflow¶
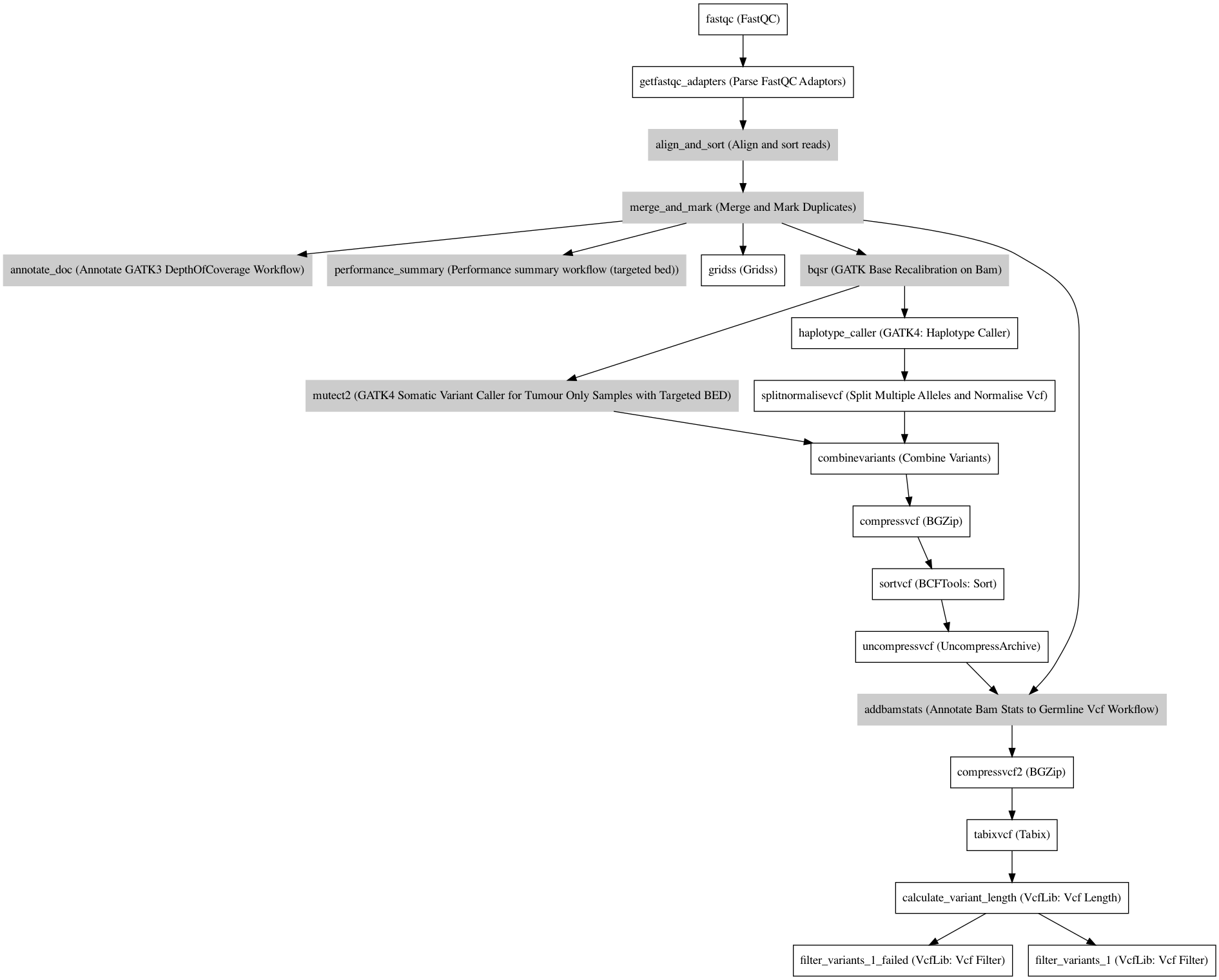
Embedded Tools¶
| FastQC | fastqc/v0.11.5 |
| Parse FastQC Adaptors | ParseFastqcAdaptors/v0.1.0 |
| Align and sort reads | BwaAligner/1.0.0 |
| Merge and Mark Duplicates | mergeAndMarkBams/4.1.3 |
| Annotate GATK3 DepthOfCoverage Workflow | AnnotateDepthOfCoverage/v0.1.0 |
| Performance summary workflow (targeted bed) | PerformanceSummaryTargeted/v0.1.0 |
| Gridss | gridss/v2.6.2 |
| GATK Base Recalibration on Bam | GATKBaseRecalBQSRWorkflow/4.1.3 |
| GATK4 Somatic Variant Caller for Tumour Only Samples with Targeted BED | GATK4_SomaticVariantCallerTumorOnlyTargeted/v0.1.1 |
| GATK4: Haplotype Caller | Gatk4HaplotypeCaller/4.1.3.0 |
| Split Multiple Alleles and Normalise Vcf | SplitMultiAlleleNormaliseVcf/v0.5772 |
| Combine Variants | combinevariants/0.0.8 |
| BGZip | bgzip/1.9 |
| BCFTools: Sort | bcftoolssort/v1.9 |
| UncompressArchive | UncompressArchive/v1.0.0 |
| Annotate Bam Stats to Germline Vcf Workflow | AddBamStatsGermline/v0.1.0 |
| Tabix | tabix/1.2.1 |
| VcfLib: Vcf Length | vcflength/v1.0.1 |
| VcfLib: Vcf Filter | vcffilter/v1.0.1 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| sample_name | String | |
| fastqs | Array<FastqGzPair> | |
| seqrun | String | SeqRun Name (for Vcf2Tsv) |
| reference | FastaWithIndexes | |
| region_bed | bed | |
| region_bed_extended | bed | |
| region_bed_annotated | bed | |
| genecoverage_bed | bed | |
| genome_file | TextFile | |
| panel_name | String | |
| vcfcols | TextFile | |
| snps_dbsnp | Gzipped<VCF> | |
| snps_1000gp | Gzipped<VCF> | |
| known_indels | Gzipped<VCF> | |
| mills_indels | Gzipped<VCF> | |
| mutalyzer_server | String | |
| pathos_db | String | |
| maxRecordsInRam | Integer | |
| gnomad | Gzipped<VCF> | |
| black_list | Optional<bed> | |
| panel_of_normals | Optional<Gzipped<VCF>> | |
| fastqc_threads | Optional<Integer> | (-t) Specifies the number of files which can be processed simultaneously. Each thread will be allocated 250MB of memory so you shouldn’t run more threads than your available memory will cope with, and not more than 6 threads on a 32 bit machine |
| align_and_sort_sortsam_tmpDir | Optional<String> | Undocumented option |
| gridss_tmpdir | Optional<String> | |
| haplotype_caller_pairHmmImplementation | Optional<String> | The PairHMM implementation to use for genotype likelihood calculations. The various implementations balance a tradeoff of accuracy and runtime. The –pair-hmm-implementation argument is an enumerated type (Implementation), which can have one of the following values: EXACT;ORIGINAL;LOGLESS_CACHING;AVX_LOGLESS_CACHING;AVX_LOGLESS_CACHING_OMP;EXPERIMENTAL_FPGA_LOGLESS_CACHING;FASTEST_AVAILABLE. Implementation: FASTEST_AVAILABLE |
| combinevariants_type | Optional<String> | germline | somatic |
| combinevariants_columns | Optional<Array<String>> | Columns to keep, seperated by space output vcf (unsorted) |
| filter_for_vcfs | Optional<String> | |
| filter_variants_1_invert | Optional<Boolean> | (-v) inverts the filter, e.g. grep -v |
Workflow Description Language¶
version development
import "tools/fastqc_v0_11_5.wdl" as F
import "tools/ParseFastqcAdaptors_v0_1_0.wdl" as P
import "tools/BwaAligner_1_0_0.wdl" as B
import "tools/mergeAndMarkBams_4_1_3.wdl" as M
import "tools/AnnotateDepthOfCoverage_v0_1_0.wdl" as A
import "tools/PerformanceSummaryTargeted_v0_1_0.wdl" as P2
import "tools/gridss_v2_6_2.wdl" as G
import "tools/GATKBaseRecalBQSRWorkflow_4_1_3.wdl" as G2
import "tools/GATK4_SomaticVariantCallerTumorOnlyTargeted_v0_1_1.wdl" as G3
import "tools/Gatk4HaplotypeCaller_4_1_3_0.wdl" as G4
import "tools/SplitMultiAlleleNormaliseVcf_v0_5772.wdl" as S
import "tools/combinevariants_0_0_8.wdl" as C
import "tools/bgzip_1_9.wdl" as B2
import "tools/bcftoolssort_v1_9.wdl" as B3
import "tools/UncompressArchive_v1_0_0.wdl" as U
import "tools/AddBamStatsGermline_v0_1_0.wdl" as A2
import "tools/tabix_1_2_1.wdl" as T
import "tools/vcflength_v1_0_1.wdl" as V
import "tools/vcffilter_v1_0_1.wdl" as V2
workflow MolpathTumorOnlyWorkflow {
input {
String sample_name
Array[Array[File]] fastqs
String seqrun
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
File region_bed
File region_bed_extended
File region_bed_annotated
File genecoverage_bed
File genome_file
String panel_name
File vcfcols
File? black_list
File snps_dbsnp
File snps_dbsnp_tbi
File snps_1000gp
File snps_1000gp_tbi
File known_indels
File known_indels_tbi
File mills_indels
File mills_indels_tbi
String mutalyzer_server
String pathos_db
Int maxRecordsInRam
File gnomad
File gnomad_tbi
File? panel_of_normals
File? panel_of_normals_tbi
Int? fastqc_threads = 4
String? align_and_sort_sortsam_tmpDir = "."
String? gridss_tmpdir = "."
String? haplotype_caller_pairHmmImplementation = "LOGLESS_CACHING"
String? combinevariants_type = "germline"
Array[String]? combinevariants_columns = ["AD", "DP", "AF", "GT"]
String? filter_for_vcfs = "length > 150"
Boolean? filter_variants_1_invert = true
}
scatter (f in fastqs) {
call F.fastqc as fastqc {
input:
reads=f,
threads=select_first([fastqc_threads, 4])
}
}
scatter (f in fastqc.datafile) {
call P.ParseFastqcAdaptors as getfastqc_adapters {
input:
fastqc_datafiles=f
}
}
scatter (Q in zip(fastqs, zip(getfastqc_adapters.adaptor_sequences, getfastqc_adapters.adaptor_sequences))) {
call B.BwaAligner as align_and_sort {
input:
sample_name=sample_name,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
fastq=Q.left,
cutadapt_adapter=Q.right.right,
cutadapt_removeMiddle3Adapter=Q.right.right,
sortsam_tmpDir=select_first([align_and_sort_sortsam_tmpDir, "."])
}
}
call M.mergeAndMarkBams as merge_and_mark {
input:
bams=align_and_sort.out,
bams_bai=align_and_sort.out_bai,
maxRecordsInRam=maxRecordsInRam,
sampleName=sample_name
}
call A.AnnotateDepthOfCoverage as annotate_doc {
input:
bam=merge_and_mark.out,
bam_bai=merge_and_mark.out_bai,
bed=region_bed_annotated,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
sample_name=sample_name
}
call P2.PerformanceSummaryTargeted as performance_summary {
input:
bam=merge_and_mark.out,
bam_bai=merge_and_mark.out_bai,
genecoverage_bed=genecoverage_bed,
region_bed=region_bed,
sample_name=sample_name,
genome_file=genome_file
}
call G.gridss as gridss {
input:
bams=[merge_and_mark.out],
bams_bai=[merge_and_mark.out_bai],
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
blacklist=black_list,
tmpdir=select_first([gridss_tmpdir, "."])
}
call G2.GATKBaseRecalBQSRWorkflow as bqsr {
input:
bam=merge_and_mark.out,
bam_bai=merge_and_mark.out_bai,
intervals=region_bed_extended,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
call G3.GATK4_SomaticVariantCallerTumorOnlyTargeted as mutect2 {
input:
bam=bqsr.out,
bam_bai=bqsr.out_bai,
intervals=region_bed_extended,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
gnomad=gnomad,
gnomad_tbi=gnomad_tbi,
panel_of_normals=panel_of_normals,
panel_of_normals_tbi=panel_of_normals_tbi
}
call G4.Gatk4HaplotypeCaller as haplotype_caller {
input:
pairHmmImplementation=select_first([haplotype_caller_pairHmmImplementation, "LOGLESS_CACHING"]),
inputRead=bqsr.out,
inputRead_bai=bqsr.out_bai,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
dbsnp=snps_dbsnp,
dbsnp_tbi=snps_dbsnp_tbi,
intervals=region_bed_extended
}
call S.SplitMultiAlleleNormaliseVcf as splitnormalisevcf {
input:
compressedVcf=haplotype_caller.out,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
call C.combinevariants as combinevariants {
input:
vcfs=[splitnormalisevcf.out, mutect2.out],
type=select_first([combinevariants_type, "germline"]),
columns=select_first([combinevariants_columns, ["AD", "DP", "AF", "GT"]])
}
call B2.bgzip as compressvcf {
input:
file=combinevariants.out
}
call B3.bcftoolssort as sortvcf {
input:
vcf=compressvcf.out
}
call U.UncompressArchive as uncompressvcf {
input:
file=sortvcf.out
}
call A2.AddBamStatsGermline as addbamstats {
input:
bam=merge_and_mark.out,
bam_bai=merge_and_mark.out_bai,
vcf=uncompressvcf.out,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
call B2.bgzip as compressvcf2 {
input:
file=addbamstats.out
}
call T.tabix as tabixvcf {
input:
inp=compressvcf2.out
}
call V.vcflength as calculate_variant_length {
input:
vcf=tabixvcf.out
}
call V2.vcffilter as filter_variants_1_failed {
input:
vcf=calculate_variant_length.out,
info_filter=select_first([filter_for_vcfs, "length > 150"])
}
call V2.vcffilter as filter_variants_1 {
input:
vcf=calculate_variant_length.out,
info_filter=select_first([filter_for_vcfs, "length > 150"]),
invert=select_first([filter_variants_1_invert, true])
}
output {
Array[Array[File]] fastq_qc = fastqc.out
File markdups_bam = merge_and_mark.out
File markdups_bam_bai = merge_and_mark.out_bai
File doc_out = annotate_doc.out
File summary = performance_summary.out
File gene_summary = performance_summary.geneFileOut
File region_summary = performance_summary.regionFileOut
File gridss_vcf = gridss.out
File gridss_bam = gridss.assembly
File haplotypecaller_vcf = haplotype_caller.out
File haplotypecaller_vcf_tbi = haplotype_caller.out_tbi
File haplotypecaller_bam = haplotype_caller.bam
File haplotypecaller_bam_bai = haplotype_caller.bam_bai
File haplotypecaller_norm = splitnormalisevcf.out
File mutect2_vcf = mutect2.variants
File mutect2_vcf_tbi = mutect2.variants_tbi
File? mutect2_bam = mutect2.out_bam
File? mutect2_bam_bai = mutect2.out_bam_bai
File mutect2_norm = mutect2.out
File addbamstats_vcf = addbamstats.out
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Molpath Tumor Only Workflow
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: ScatterFeatureRequirement
- class: SubworkflowFeatureRequirement
- class: MultipleInputFeatureRequirement
inputs:
- id: sample_name
type: string
- id: fastqs
type:
type: array
items:
type: array
items: File
- id: seqrun
doc: SeqRun Name (for Vcf2Tsv)
type: string
- id: reference
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: region_bed
type: File
- id: region_bed_extended
type: File
- id: region_bed_annotated
type: File
- id: genecoverage_bed
type: File
- id: genome_file
type: File
- id: panel_name
type: string
- id: vcfcols
type: File
- id: black_list
type:
- File
- 'null'
- id: snps_dbsnp
type: File
secondaryFiles:
- pattern: .tbi
- id: snps_1000gp
type: File
secondaryFiles:
- pattern: .tbi
- id: known_indels
type: File
secondaryFiles:
- pattern: .tbi
- id: mills_indels
type: File
secondaryFiles:
- pattern: .tbi
- id: mutalyzer_server
type: string
- id: pathos_db
type: string
- id: maxRecordsInRam
type: int
- id: gnomad
type: File
secondaryFiles:
- pattern: .tbi
- id: panel_of_normals
type:
- File
- 'null'
secondaryFiles:
- pattern: .tbi
- id: fastqc_threads
doc: |-
(-t) Specifies the number of files which can be processed simultaneously. Each thread will be allocated 250MB of memory so you shouldn't run more threads than your available memory will cope with, and not more than 6 threads on a 32 bit machine
type: int
default: 4
- id: align_and_sort_sortsam_tmpDir
doc: Undocumented option
type: string
default: .
- id: gridss_tmpdir
type: string
default: .
- id: haplotype_caller_pairHmmImplementation
doc: |-
The PairHMM implementation to use for genotype likelihood calculations. The various implementations balance a tradeoff of accuracy and runtime. The --pair-hmm-implementation argument is an enumerated type (Implementation), which can have one of the following values: EXACT;ORIGINAL;LOGLESS_CACHING;AVX_LOGLESS_CACHING;AVX_LOGLESS_CACHING_OMP;EXPERIMENTAL_FPGA_LOGLESS_CACHING;FASTEST_AVAILABLE. Implementation: FASTEST_AVAILABLE
type: string
default: LOGLESS_CACHING
- id: combinevariants_type
doc: germline | somatic
type: string
default: germline
- id: combinevariants_columns
doc: Columns to keep, seperated by space output vcf (unsorted)
type:
type: array
items: string
default:
- AD
- DP
- AF
- GT
- id: filter_for_vcfs
type: string
default: length > 150
- id: filter_variants_1_invert
doc: (-v) inverts the filter, e.g. grep -v
type: boolean
default: true
outputs:
- id: fastq_qc
type:
type: array
items:
type: array
items: File
outputSource: fastqc/out
- id: markdups_bam
type: File
secondaryFiles:
- pattern: .bai
outputSource: merge_and_mark/out
- id: doc_out
type: File
outputSource: annotate_doc/out
- id: summary
type: File
outputSource: performance_summary/out
- id: gene_summary
type: File
outputSource: performance_summary/geneFileOut
- id: region_summary
type: File
outputSource: performance_summary/regionFileOut
- id: gridss_vcf
type: File
outputSource: gridss/out
- id: gridss_bam
type: File
outputSource: gridss/assembly
- id: haplotypecaller_vcf
type: File
secondaryFiles:
- pattern: .tbi
outputSource: haplotype_caller/out
- id: haplotypecaller_bam
type: File
secondaryFiles:
- pattern: .bai
outputSource: haplotype_caller/bam
- id: haplotypecaller_norm
type: File
outputSource: splitnormalisevcf/out
- id: mutect2_vcf
type: File
secondaryFiles:
- pattern: .tbi
outputSource: mutect2/variants
- id: mutect2_bam
type:
- File
- 'null'
secondaryFiles:
- pattern: .bai
outputSource: mutect2/out_bam
- id: mutect2_norm
type: File
outputSource: mutect2/out
- id: addbamstats_vcf
type: File
outputSource: addbamstats/out
steps:
- id: fastqc
label: FastQC
in:
- id: reads
source: fastqs
- id: threads
source: fastqc_threads
scatter:
- reads
run: tools/fastqc_v0_11_5.cwl
out:
- id: out
- id: datafile
- id: getfastqc_adapters
label: Parse FastQC Adaptors
in:
- id: fastqc_datafiles
source: fastqc/datafile
scatter:
- fastqc_datafiles
run: tools/ParseFastqcAdaptors_v0_1_0.cwl
out:
- id: adaptor_sequences
- id: align_and_sort
label: Align and sort reads
in:
- id: sample_name
source: sample_name
- id: reference
source: reference
- id: fastq
source: fastqs
- id: cutadapt_adapter
source: getfastqc_adapters/adaptor_sequences
- id: cutadapt_removeMiddle3Adapter
source: getfastqc_adapters/adaptor_sequences
- id: sortsam_tmpDir
source: align_and_sort_sortsam_tmpDir
scatter:
- fastq
- cutadapt_adapter
- cutadapt_removeMiddle3Adapter
scatterMethod: dotproduct
run: tools/BwaAligner_1_0_0.cwl
out:
- id: out
- id: merge_and_mark
label: Merge and Mark Duplicates
in:
- id: bams
source: align_and_sort/out
- id: maxRecordsInRam
source: maxRecordsInRam
- id: sampleName
source: sample_name
run: tools/mergeAndMarkBams_4_1_3.cwl
out:
- id: out
- id: annotate_doc
label: Annotate GATK3 DepthOfCoverage Workflow
in:
- id: bam
source: merge_and_mark/out
- id: bed
source: region_bed_annotated
- id: reference
source: reference
- id: sample_name
source: sample_name
run: tools/AnnotateDepthOfCoverage_v0_1_0.cwl
out:
- id: out
- id: out_sample_summary
- id: performance_summary
label: Performance summary workflow (targeted bed)
in:
- id: bam
source: merge_and_mark/out
- id: genecoverage_bed
source: genecoverage_bed
- id: region_bed
source: region_bed
- id: sample_name
source: sample_name
- id: genome_file
source: genome_file
run: tools/PerformanceSummaryTargeted_v0_1_0.cwl
out:
- id: out
- id: geneFileOut
- id: regionFileOut
- id: gridss
label: Gridss
in:
- id: bams
source:
- merge_and_mark/out
linkMerge: merge_nested
- id: reference
source: reference
- id: blacklist
source: black_list
- id: tmpdir
source: gridss_tmpdir
run: tools/gridss_v2_6_2.cwl
out:
- id: out
- id: assembly
- id: bqsr
label: GATK Base Recalibration on Bam
in:
- id: bam
source: merge_and_mark/out
- id: intervals
source: region_bed_extended
- id: reference
source: reference
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
run: tools/GATKBaseRecalBQSRWorkflow_4_1_3.cwl
out:
- id: out
- id: mutect2
label: GATK4 Somatic Variant Caller for Tumour Only Samples with Targeted BED
in:
- id: bam
source: bqsr/out
- id: intervals
source: region_bed_extended
- id: reference
source: reference
- id: gnomad
source: gnomad
- id: panel_of_normals
source: panel_of_normals
run: tools/GATK4_SomaticVariantCallerTumorOnlyTargeted_v0_1_1.cwl
out:
- id: variants
- id: out_bam
- id: out
- id: haplotype_caller
label: 'GATK4: Haplotype Caller'
in:
- id: pairHmmImplementation
source: haplotype_caller_pairHmmImplementation
- id: inputRead
source: bqsr/out
- id: reference
source: reference
- id: dbsnp
source: snps_dbsnp
- id: intervals
source: region_bed_extended
run: tools/Gatk4HaplotypeCaller_4_1_3_0.cwl
out:
- id: out
- id: bam
- id: splitnormalisevcf
label: Split Multiple Alleles and Normalise Vcf
in:
- id: compressedVcf
source: haplotype_caller/out
- id: reference
source: reference
run: tools/SplitMultiAlleleNormaliseVcf_v0_5772.cwl
out:
- id: out
- id: combinevariants
label: Combine Variants
in:
- id: vcfs
source:
- splitnormalisevcf/out
- mutect2/out
- id: type
source: combinevariants_type
- id: columns
source: combinevariants_columns
run: tools/combinevariants_0_0_8.cwl
out:
- id: out
- id: compressvcf
label: BGZip
in:
- id: file
source: combinevariants/out
run: tools/bgzip_1_9.cwl
out:
- id: out
- id: sortvcf
label: 'BCFTools: Sort'
in:
- id: vcf
source: compressvcf/out
run: tools/bcftoolssort_v1_9.cwl
out:
- id: out
- id: uncompressvcf
label: UncompressArchive
in:
- id: file
source: sortvcf/out
run: tools/UncompressArchive_v1_0_0.cwl
out:
- id: out
- id: addbamstats
label: Annotate Bam Stats to Germline Vcf Workflow
in:
- id: bam
source: merge_and_mark/out
- id: vcf
source: uncompressvcf/out
- id: reference
source: reference
run: tools/AddBamStatsGermline_v0_1_0.cwl
out:
- id: out
- id: compressvcf2
label: BGZip
in:
- id: file
source: addbamstats/out
run: tools/bgzip_1_9.cwl
out:
- id: out
- id: tabixvcf
label: Tabix
in:
- id: inp
source: compressvcf2/out
run: tools/tabix_1_2_1.cwl
out:
- id: out
- id: calculate_variant_length
label: 'VcfLib: Vcf Length'
doc: Add the length column for the output of AddBamStats
in:
- id: vcf
source: tabixvcf/out
run: tools/vcflength_v1_0_1.cwl
out:
- id: out
- id: filter_variants_1_failed
label: 'VcfLib: Vcf Filter'
in:
- id: vcf
source: calculate_variant_length/out
- id: info_filter
source: filter_for_vcfs
run: tools/vcffilter_v1_0_1.cwl
out:
- id: out
- id: filter_variants_1
label: 'VcfLib: Vcf Filter'
in:
- id: vcf
source: calculate_variant_length/out
- id: info_filter
source: filter_for_vcfs
- id: invert
source: filter_variants_1_invert
run: tools/vcffilter_v1_0_1.cwl
out:
- id: out
id: MolpathTumorOnlyWorkflow